- You are using the MALDI-TOF1 Biotyper to identify bacterial isolates
- You want to select representative isolates for further experiments
- You need fast and automated selection decisions that you can retrace
maldipickr provides documented and tested R functions that will help you dereplicate MALDI-TOF data and cherry-pick representative spectra of microbial isolates.
Graphical overview
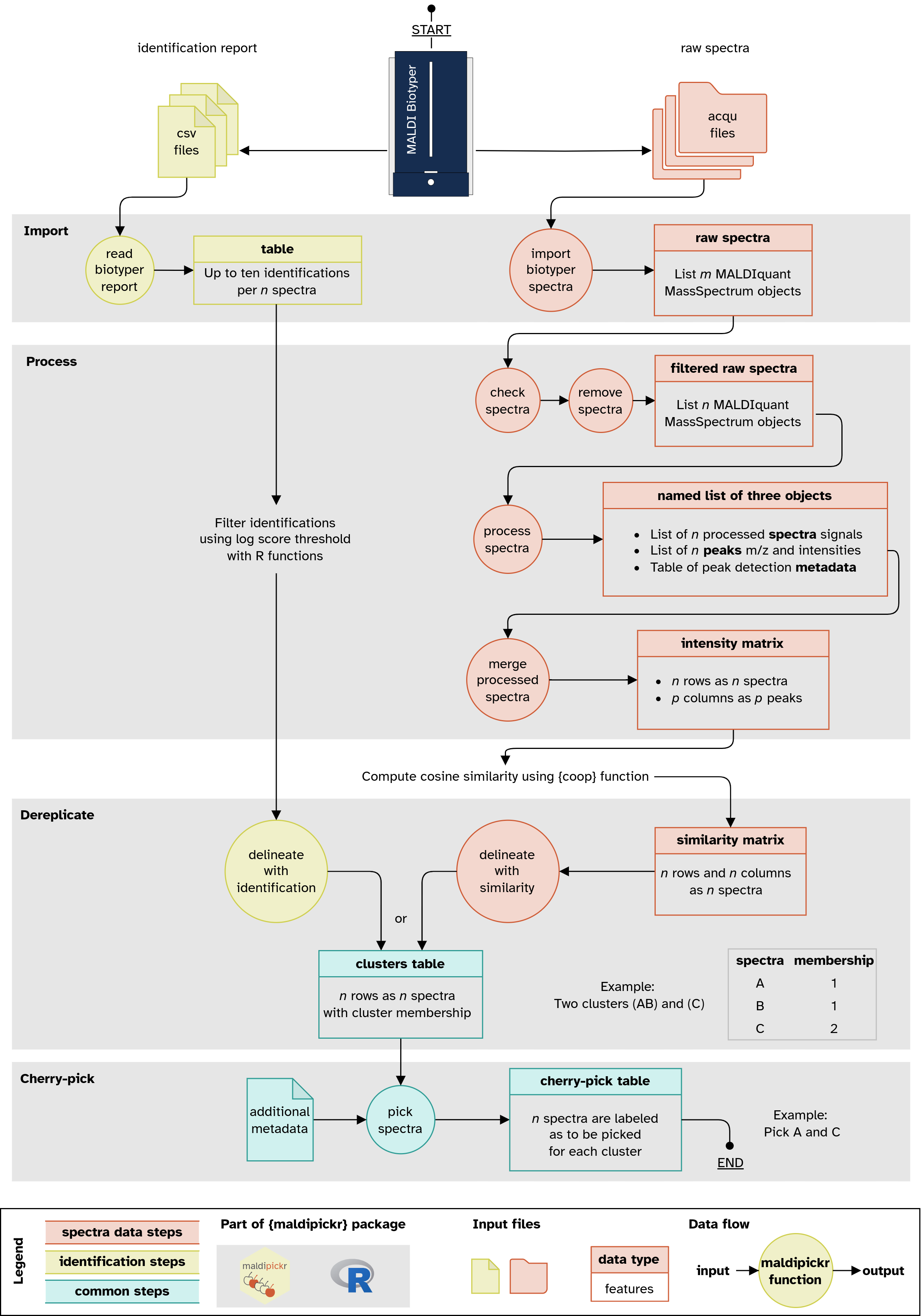
Illustration (click for a bigger version) of the data flow when using maldipickr to cherry-pick bacterial isolates with MALDI Biotyper. It depicts the two possible approaches using either taxonomic identification reports (left) or spectra data (right).
Quickstart
How to cherry-pick bacterial isolates with MALDI Biotyper:
Using taxonomic identification report
library(maldipickr)
# Import Biotyper CSV report
# and glimpse at the table
report_tbl <- read_biotyper_report(
system.file("biotyper_unknown.csv", package = "maldipickr")
)
report_tbl %>%
dplyr::select(name, bruker_species, bruker_log)
#> # A tibble: 4 × 3
#> name bruker_species bruker_log
#> <chr> <chr> <dbl>
#> 1 unknown_isolate_1 not reliable identification 1.33
#> 2 unknown_isolate_2 not reliable identification 1.4
#> 3 unknown_isolate_3 Faecalibacterium prausnitzii 1.96
#> 4 unknown_isolate_4 Faecalibacterium prausnitzii 2.07
# Delineate clusters from the identifications after filtering the reliable ones
# and cherry-pick one representative spectra.
# The chosen ones are indicated by `to_pick` column
report_tbl <- report_tbl %>%
dplyr::mutate(
bruker_species = dplyr::if_else(bruker_log >= 2, bruker_species,
"not reliable identification")
)
report_tbl %>%
delineate_with_identification() %>%
pick_spectra(report_tbl, criteria_column = "bruker_log") %>%
dplyr::relocate(name, to_pick, bruker_species)
#> Generating clusters from single report
#> # A tibble: 4 × 11
#> name to_pick bruker_species membership cluster_size sample_name hit_rank
#> <chr> <lgl> <chr> <int> <int> <chr> <int>
#> 1 unknown_i… TRUE not reliable … 2 1 <NA> 1
#> 2 unknown_i… TRUE not reliable … 3 1 <NA> 1
#> 3 unknown_i… TRUE not reliable … 4 1 <NA> 1
#> 4 unknown_i… TRUE Faecalibacter… 1 1 <NA> 1
#> # ℹ 4 more variables: bruker_quality <chr>, bruker_taxid <dbl>,
#> # bruker_hash <chr>, bruker_log <dbl>Using spectra data
library(maldipickr)
# Set up the directory location of your spectra data
spectra_dir <- system.file("toy-species-spectra", package = "maldipickr")
# Import and process the spectra
processed <- spectra_dir %>%
import_biotyper_spectra() %>%
process_spectra()
# Delineate spectra clusters using Cosine similarity
# and cherry-pick one representative spectra.
# The chosen ones are indicated by `to_pick` column
processed %>%
list() %>%
merge_processed_spectra() %>%
coop::tcosine() %>%
delineate_with_similarity(threshold = 0.92) %>%
set_reference_spectra(processed$metadata) %>%
pick_spectra() %>%
dplyr::relocate(name, to_pick)
#> # A tibble: 6 × 7
#> name to_pick membership cluster_size SNR peaks is_reference
#> <chr> <lgl> <int> <int> <dbl> <int> <lgl>
#> 1 species1_G2 FALSE 1 4 5.09 21 FALSE
#> 2 species2_E11 FALSE 2 2 5.54 22 FALSE
#> 3 species2_E12 TRUE 2 2 5.63 23 TRUE
#> 4 species3_F7 FALSE 1 4 4.89 26 FALSE
#> 5 species3_F8 TRUE 1 4 5.56 25 TRUE
#> 6 species3_F9 FALSE 1 4 5.40 25 FALSEInstallation
maldipickr is available on the CRAN and on GitHub.
To install the latest CRAN release, use the following command in R:
install.packages("maldipickr")To install the development version, use the following command in R:
remotes::install_github("ClavelLab/maldipickr", build_vignettes = TRUE)Troubleshoot
If something unexpected happened when using this package, please first search the current open or closed issues to look for similar problems. If you are the first, you are more than welcome to open a new issue using the “Bug report” template with a minimal reprex.
Contribute
All contributions are welcome and the CONTRIBUTING.md documents how to participate.
Please note that the {maldipickr} package is released with a Contributor Code of Conduct. By contributing to this project, you agree to abide by its terms.
Credits
Acknowledgements
This R package is developed for spectra data generated by the Bruker MALDI Biotyper device. The {maldipickr} package is built from a suite of Rmarkdown files using the {fusen} package by Rochette S (2023). It relies on:
- the
{MALDIquant}package from Gibb & Strimmer (2012) for spectra functions - the work of Strejcek et al. (2018) for the dereplication procedure.
Disclaimer
The developers of this package are part of the Clavel Lab and are not affiliated with the company Bruker, therefore this package is independent of the company and is distributed under the GPL-3.0 License.
The hexagonal logo was created by Charlie Pauvert and uses the Atkinson Hyperlegible font font and a color palette generated at coolors.co.
References
- Gibb S & Strimmer K (2012). “MALDIquant: a versatile R package for the analysis of mass spectrometry data”. Bioinformatics 28, 2270-2271. https://doi.org/10.1093/bioinformatics/bts447.
- Rochette S (2023). “fusen: Build a Package from Rmarkdown Files”. https://thinkr-open.github.io/fusen/, https://github.com/Thinkr-open/fusen.
- Strejcek M, Smrhova T, Junkova P & Uhlik O (2018). “Whole-Cell MALDI-TOF MS versus 16S rRNA Gene Analysis for Identification and Dereplication of Recurrent Bacterial Isolates.” Frontiers in Microbiology 9 https://doi.org/10.3389/fmicb.2018.01294.